
Warenkorb
Es gibt keine Artikel mehr in Ihrem Warenkorb





{kind=link}
{kind=link}
{kind=link}
Kongenitale Pulmonalstenose - Erler Zimmer 3D Anatomie Serie MP2034
erler zimmer
EZ-MP2034
632,08 €
Bruttopreis
Hergestellt im ultrahochauflösenden 3D-Druck in voller Farbe.
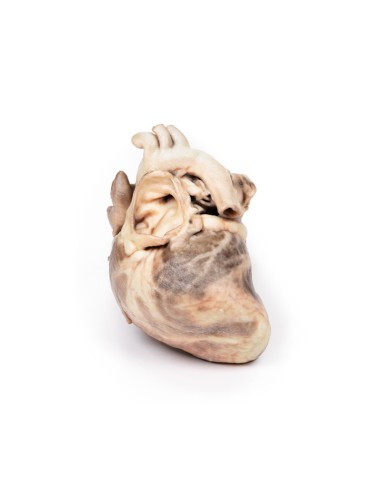
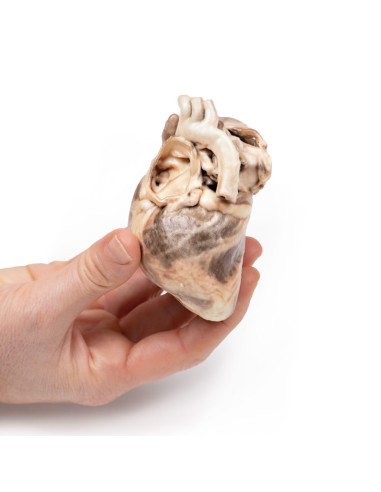
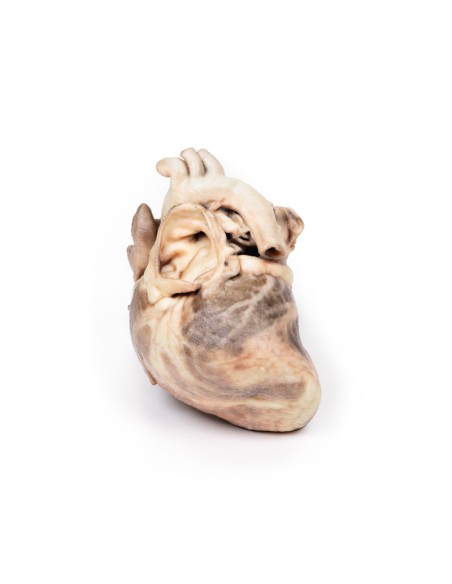
Kongenitale Pulmonalstenose - Erler Zimmer 3D-Anatomie Serie MP2034
Dieses Präparationsmodell der kongenitalen Pulmonalstenose ist Teil der exklusiven Monash 3D-Anatomie-Serie, einer umfassenden Serie von menschlichen Präparaten, die mit ultrahochauflösendem 3D-Farbdruck reproduziert wurden.
Klinische Vorgeschichte
Bei diesem männlichen Kind wurde bei der Geburt ein Herzgeräusch entdeckt. Bis zum Alter von 10 Monaten blieb er gesund. Zwei Wochen vor der Krankenhauseinweisung wurde er träge, entwickelte einen leichten Husten und nahm plötzlich stark an Gewicht zu. Zehn Tage vor der Einlieferung entwickelte er Schwellungen an Händen, Füßen und im Gesicht. Bei der Untersuchung war er ein pummeliges Baby mit einem Blutdruck von 90/59 mmHg. Das gesamte Präkordium war stark zittrig, und in der Lungengegend war ein maximales systolisches Geräusch zu hören. Im Gesicht und an den Beinen traten Ödeme auf. Nach der Einlieferung nahm das Kind weiter an Gewicht zu, ohne dass es auf die Behandlung ansprach. Der Tod wurde durch kongestives Herzversagen verursacht.
Pathologie
Bei dem Präparat handelt es sich um das Herz des Kindes. Bei der Ansicht von der linken Seite ist zu beachten, dass die Pulmonalarterie geöffnet wurde, um die Oberseite der Pulmonalklappe sichtbar zu machen. Diese abnorme Klappe besteht aus einem verdickten konischen Diaphragma mit einer Öffnung von 2 mm Durchmesser am Apex. Die offene Pulmonalarterie weist eine große poststenotische Dilatation auf. Die Vergrößerung des rechten Herzens ist auf eine ausgeprägte Dilatation des rechten Vorhofs und des rechten Vorhofs (offen) sowie auf eine Hypertrophie des rechten Ventrikels zurückzuführen (die Wand wurde an zwei Stellen durchtrennt [eine durchdringend, eine nicht], um das hypertrophierte Myokard freizulegen). Es handelt sich um eine reine Pulmonalklappenstenose.
Weitere Informationen
Die Pulmonalstenose macht etwa 7 % der angeborenen Herzfehler aus. Sie kann jedoch auch im späteren Leben als Folge eines erhöhten Lungendrucks auftreten. Bei Säuglingen kann sie als isolierter Defekt oder als Teil einer komplexeren Läsion, wie der Fallot-Tetralogie, auftreten.
Letztere ist eine Kombination aus vier angeborenen Anomalien, darunter ein Ventrikelseptumdefekt, eine Pulmonalklappenstenose, eine verlegte Aorta und eine Hypertrophie des rechten Ventrikels. Anomalien, die diesem Exemplar ähneln, können heute chirurgisch korrigiert werden.
Der natürliche Verlauf der kongenitalen Pulmonalstenose variiert je nach Alter und Schweregrad im Entwicklungsalter. Die Pulmonalstenose kann auf einer Skala von leicht, mittelschwer und schwer eingestuft werden. Während eine leichte Pulmonalstenose über mehrere Jahre hinweg asymptomatisch und indolent bleiben kann, ohne dass eine Diagnose gestellt wird, kann eine mittelschwere Pulmonalstenose rasch zu einer schweren Stenose fortschreiten. Eine schwere Pulmonalstenose hat aufgrund des erhöhten Drucks, der zu einer rechtsventrikulären Hypertrophie führt, erhebliche Auswirkungen auf das Herz. Eine schwere Pulmonalstenose geht häufig mit einer Abflussbehinderung des rechten Ventrikels und anschließendem Herzversagen einher. Daher kann sich eine mittelschwere bis schwere Pulmonalstenose als Dyspnoe, Brustschmerzen oder Synkope äußern.
Zu den weiteren Komplikationen einer schweren Pulmonalstenose gehören infektiöse Endokarditis und Herzrhythmusstörungen, die durch Umbau und Vernarbung der Ventrikel- und Vorhofwände verursacht werden. Die Diagnose einer Pulmonalstenose wird häufig mittels transthorakaler Echokardiographie gestellt. Weitere bildgebende Verfahren sind MRT und CT.
Welche Vorteile bietet die Sammlung anatomischer Präparate der Monash University gegenüber Plastikmodellen oder plastifizierten menschlichen Präparaten?
- Jede Körpernachbildung wurde sorgfältig aus ausgewählten Röntgendaten von Patienten oder menschlichen Kadavern erstellt, die von einem hochqualifizierten Team von Anatomen am Monash University Centre for Human Anatomy Education ausgewählt wurden, um eine Reihe von klinisch wichtigen Bereichen der Anatomie mit einer Qualität und Genauigkeit zu veranschaulichen, die mit herkömmlichen anatomischen Modellen nicht erreicht werden kann - dies ist echte Anatomie, keine stilisierte Anatomie.
- Jede Körpernachbildung wurde von einem Team hochqualifizierter Anatomen am Centre for Human Anatomy Education der Monash University genauestens überprüft, um die anatomische Genauigkeit des Endprodukts zu gewährleisten.
- Die Körpernachbildungen sind kein echtes menschliches Gewebe und unterliegen daher keinen Transport-, Einfuhr- oder Verwendungsbeschränkungen in Bildungseinrichtungen, die nicht über eine Anatomielizenz verfügen. Die Monash 3D Anatomy Dissection Serie vermeidet diese und andere ethische Probleme, die beim Umgang mit plastifizierten menschlichen Überresten auftreten.
No reviews
Tap to zoom
